_Speciation_vs_chromosome_evolution_vertebrates.png)
THERYA, 2025, Vol. 16(1):5-12 DOI:10.12933/therya-25-6184 ISSN 2007-3364
Chromosome evolution and speciation:
Revisiting Bush et al. (1977)
Craig Moritz1*, and Sally Potter2
1 Research School of Biology, The Australian National University, ACT 2601, Australia. Email: craig.moritz@anu.edu.au (CM).
2 School of Natural Sciences, Macquarie University, Macquarie Park, NSW 2901, Australia. Email: sally.potter@mq.edu.au (SP).
* Corresponding author: https://orcid.org/0000-0001-5313-7279.
Whether and how chromosome change can drive speciation has long attracted the interest of evolutionary biologists. In a seminal paper, Bush et al. (1977) demonstrated wide variation in rates of macroscopic chromosome change across vertebrates and a positive correlation with rates of speciation at genus-level, and then considered possible causal processes. We revisit the key findings of this paper, highlighting how rapidly advancing knowledge of genome organisation and function shed new light on this long standing question. The central findings of Bush et al. (1977) have endured. There is now great opportunity to apply new genome technologies to understand the interaction of genome change and function in the classic systems of chromosomal speciation discovered through classical cytogenetics.
El hecho de si el cambio cromosómico puede impulsar la especiación y cómo puede hacerlo ha atraído desde hace tiempo el interés de los biólogos evolutivos. En un artículo fundamental, Bush et al. (1977) demostraron una amplia variación en las tasas de cambio cromosómico macroscópico en vertebrados y una correlación positiva con las tasas de especiación a nivel de género, y luego consideraron los posibles procesos causales. Repasamos los hallazgos clave de este artículo, destacando cómo el rápido avance del conocimiento de la organización y función del genoma arroja nueva luz sobre esta antigua pregunta. Los hallazgos centrales de Bush et al. (1977) han perdurado. Ahora existe una gran oportunidad de aplicar nuevas tecnologías genómicas para comprender la interacción del cambio y la función del genoma en los sistemas clásicos de especiación cromosómica descubiertos mediante la citogenética clásica.
Keywords: Chromosomal speciation; genome evolution; cytogenetics.
© 2025 Asociación Mexicana de Mastozoología, www.mastozoologiamexicana.org
Introduction
Jim Patton, chromosomes and speciation. Jim Patton is an exemplary scholar and educator of all things relating to diversity of mammals. He is also a selfless mentor to early career researchers, including the lead author from the outset of his PhD, throughout his postdoctoral studies, and then as a faculty colleague. His seminal contributions are many and varied, including works on systematics, adaptation and speciation with a special focus on rodents, the most speciose order of mammals. His earliest works demonstrated the extraordinary diversity of chromosome number and form among rodents (e. g. Patton 1967) leading to many papers exploring how genome reorganisation contributed to speciation. Here we focus on one highly influential paper, Bush et al. (1977), on which Patton was senior author. We first summarise the context and key points of this paper. Then, some 47 years on, we revisit the key findings from the perspectives of new evidence on genome restructuring in mammals and also new theory.
The era of “classical cytogenetics” revealed substantial variation in chromosome organisation –differences in number of entire chromosomes and of chromosome arms– among closely related species. This, together with observations of disrupted meiosis in hybrids, quite naturally led to the conclusion that chromosome change could directly cause speciation (summarised in White 1978). At the same time, there was growing acceptance that genetic drift in small populations increases the likelihood of speciation – Wright’s (1931, 1940) Shifting Balance Theory.
In this context, Bush et al. (1977) set out to test the prediction that rates of speciation across representative genera of vertebrates should be correlated with rates of chromosome change. The authors then linked differences in rates to ecological attributes that should lead to strong genetic drift. The key results (Figure1) were that: i) rates of both speciation and chromosome change are higher in mammals than in other classes of vertebrates; ii) across vertebrate genera, there is a strong correlation between these rates; and iii) mammalian genera with high rates of chromosome change do have ecological characteristics that can be expected to enhance drift. This quantitative analysis, together with a parallel finding for plants (Levin and Wilson 1976) was a seminal contribution to the developing theory of speciation by chromosome change. Bush et al. (1977) noted that correlation does not prove that chromosome changes can directly cause speciation, but they did point out several ways by which this might happen, including reduced fitness of hybrids, changes in gene regulation and creation of novel linkage groups.
In subsequent papers (Patton and Sherwood 1983; Patton 2004), Patton was less enamoured with the hypothesis that meiotic dysfunction was the link between chromosome change and speciation due to the dependence on strong genetic drift and increasing observations of within-species polymorphism for the same forms of chromosome change as were supposed to cause speciation (see also Sites and Moritz 1987; Coyne and Orr 2004). One exception is where multiple independent fusions establish in separate populations (via neutral processes) yet cause severe meiotic problems in hybrids if particular arms are involved in different fusions. This is called “monobrachial homology” and is a form of Dobzhansky-Muller incompatibility (Baker and Bickham 1986). Instead, Patton (2004) highlighted new theory and evidence (Navarro and Barton 2003) pointing to effects of chromosome change on the genomic distribution of recombination, with subsequent effects on accumulation of adaptive mutations within species and incompatibilities between species (see also Kirkpatrick and Barton 2006).
As Patton (2004) noted, the emerging ability to compare high-quality, de-novo whole genome assemblies reveals much more extensive chromosome change than was evident from numbers of chromosomes or chromosome arms that formed the basis of the analyses in Bush et al. (1977). Following his example, humans (2n = 46) and the other great apes (2n = 48) differ by just one fusion, yet recent genome comparisons reveal >17,000 fixed differences for structural variants (insertions, deletions and inversions) spanning (>18Mb) in humans on the branch from our common ancestor with chimpanzees, of which many are associated with changes in gene expression (Kronenberg et al. 2018). High-resolution analyses of copy number variants are providing important insights into genome change and speciation. For example, expansions of X-linked gene families associated with spermatogenesis (“ampliconic gene families”) have an important role in reproductive isolation in apes and rodents (Kopiana et al. 2022).
Assessment of key insights from Bush et al. (1977)
Do mammals have a higher rate of chromosome change and is that correlated with a higher rate of speciation and with small population size? Recent compilations of cytogenetically characterised chromosome variation in mammals support the main findings of Bush et al. (1977). Using much more extensive data and phylogenetic – macroevolutionary comparisons, Martinez et al. (2017) confirmed wide (two orders of magnitude) variation in rates of macro-chromosomal change among families of mammals and associations of high rates of chromosome change with geographic range and litter size. Using data from Martinez et al. (2017) and other sources, Herrick and Sclavi (2019) confirm a strong association between rates of chromosome change and speciation across orders and families of mammals. Interestingly, there was no correlation between degree of conservation of syntenic gene blocks and rates of chromosome change or speciation. Within diprotodontian marsupials, Westerman et al. (2010) contrasted strong conservation of the ancestral karyotype in several families with extensive chromosome change including fusion, fissions, centric shifts, in others, e. g. Macropodidae. A particular form of intrachromosomal rearrangement, centromere repositioning, occurs when neocentromeres establish to replace ancestral ones and is common in several cytogenetically variable mammalian taxa (reviewed in Brannan et al. 2024). There has been less attention to indicators of small population size, though for Carnivora, Jonicka et al. (2024) found that species with smaller ranges have a higher rate of Robertsonian translocations and small population size has been associated with higher rates of chromosomal rearrangement fixation in some groups (e. g. Australian rock-wallabies –Petrogale– see below). However, our ability to test this prediction and tease apart the role of genetic drift from selection remains a challenge.
The ongoing revolution in long-read sequencing has greatly improved the capacity to produce highly contiguous “chromosome scale” assemblies of genomes and so detect chromosome change in exquisite detail (Figure 2). The addition of HiC chromatin contact maps enables assembly of long-read scaffolds (because linked segments have more interactions) and, along with other methods (summarised in Mohan et al. 2024), reveals structure of chromatin in the interphase nucleus, including active (A, “euchromatin”) or inactive (B, “heterochromatin”) compartments and topologically associated domains (TADs, regions of coordinated gene expression). When combined with efficient algorithms to estimate ancestral genome arrangements (e. g. Muffato et al. 2023; Yu et al. 2024), comparing whole-genome assemblies provides rich, highly-resolved information on conserved syntenic regions, Evolutionary Breakpoint Regions (EBRs) at the boundaries of these regions and genome reshuffling at macroevolutionary scale. What does this rapidly accelerating knowledge tell us about the nature, cause and effects of chromosome rearrangements in mammals?
Comparisons of genome assemblies across vertebrates confirm the extraordinarily high rate of chromosome rearrangement in therian mammals relative to other amniotes – amphibians, birds, and non-flying reptiles (Waters et al. 2021; Bredeson et al. 2024). In a recent review, Damas et al. (2021) conclude that mammals are more prone to interchromosomal rearrangements and birds to intrachromosomal change. In a separate comparison of 92 vertebrate genome assemblies, including ancestral reconstructions, Muffato et al. (2023) again found that mammals have an elevated rate of interchromosomal rearrangements, with especially high rates in gibbons, dogs and murid rodents. They also inferred that rates of intrachromosomal rearrangements were similar across mammals and saurian reptiles, but higher in teleosts perhaps due to diploidisation in the latter.
Focussing on artiodactyls (e. g., ungulates), another group for which Bush et al. (1977) reported a high rate of chromosome change, comparisons of whole genome assemblies found inversions to be the most common form of chromosome rearrangement, with fusions/fissions mostly occurring in ancestral lineages (Aria-Sarda et al. 2023). Muntjaks are a special case, with chromosome numbers varying from 2n = 46 to 2n = 6/7. Based on high-quality genome assemblies, Yin et al. (2021) estimated 31 fusion events (28 tandem and 3 Robertsonian) over the past ~ 2 My, but did not detect any increase in SNP divergence rates compared to Bovids with fewer chromosome rearrangements. Interestingly, the inferred pulse of fusions at ~ 1Myr corresponded with a period of reduced population size in the species with low chromosome numbers.
Do chromosome changes cause speciation? Bush et al. (1977; also White 1978; King 1993) summarised multiple ways by which chromosome rearrangements could initiate speciation. Recent reviews (Berdan et al. 2023; Lucek et al. 2023) support multiple effects –a given chromosome rearrangement can contribute to reproductive isolation and speciation via multiple, non-exclusive mechanisms– reduction in hybrid fitness (underdominance), change in recombination rate, and changes in gene expression. Meiotic drive can also contribute to establishment of chromosome rearrangements within populations. In parallel, there has been long-standing debate about whether chromosomal rearrangements that cause speciation always arise in isolated or allopatric populations, or can established in the face of gene flow and within the range of the ancestral form (reviewed in White 1978, King 1993). Most theory and evidence support allopatric origins of derived chromosome forms, though it is plausible that, via suppression of recombination, inversions could enhance adaptive speciation in parapatry. In contrast, there is little support for White’s “Stasipatric” speciation model in which underdominant chromosome changes establish in sympatry because of increased fitness of homozygotes.
Underdominance. There is still limited evidence supporting strong underdominance due to single fusions or inversions as such mutations are unlikely to establish and/or are eliminated during gametogenesis (cells with univalents failing pachytene-diplotene transition or, for unbalanced products of recombination, shunted to polar bodies in oogenesis; Berdan et al. 2023). One exception is X-autosome translocations which cause substantial male sterility (Ashley 2002). However, heterozygotes for multiple chromosome rearrangements, as might accumulate via neutral processes in allopatry, can have substantially reduced fitness (Baker and Bickham 1986). Rock-wallabies are one example, in which such “monobrachial homology” occurs between some parapatric taxa and is expected to substantially reduce fitness of hybrids. The east coast penicillata group consists of eight parapatric and allopatric species which all have varied chromosome numbers due to Robertsonian fusions. Experimental crosses between species highlighted reduced fertility, which led to the description of three new species (Eldridge and Close 1992; Close et al. 1996). Haldane’s Rule (Haldane 1922), where males are sterile and females are sub-fertile, is associated with divergence of species in this group and highlight potential mechanisms of a role of the sex chromosomes or meiotic drive in reproductive isolation. These organisms are restricted to complex rocky outcrops and have small population sizes, so it is thought chromosomal rearrangements could be fixed due to genetic drift alone. Theory would predict that if underdominance is the driver of divergence in such a system, rearranged and non-rearranged regions of the genome would have comparable levels of genetic divergence. In contrast, if recombination suppression was the driving force for reproductive isolation, then we would expect the rearranged regions to have greater divergence than non-rearranged regions. Comparison of exonic sequences revealed higher sequence divergence on the X chromosome compared to autosomes, and differences between rearranged and non-rearranged regions of the genome, but not in the expected direction (Potter et al. 2022). Stronger tests of this hypothesis require full genome comparisons, using long-read sequencing, which is underway.
Chromosome rearrangements and recombination. There is now abundant evidence for suppression of recombination in polymorphic inversions within species and increased divergence between species or ecotypes in rearranged versus co-linear regions (Wellenreuther and Bernatchez 2018). For example, genes linked within a 41 Mb polymorphic inversion are responsible for multi-trait divergence between forest and prairie ecotypes of deer mice (Hager et al. 2022). In human-chimpanzee comparisons, recombination is reduced in segments spanning eight major inversion differences (Farre et al. 2012). Sequence divergence in inversions, which often span multiple genes, can be attributed to several, non-exclusive processes. These include adaptive divergence and accumulation of deleterious alleles, both of which are expected to occur over the life history of an inversion (Faria et al. 2018; Berdan et al. 2023), and each of which can lead to Dobzhansky-Mueller incompatibilities (Kirkpatrick and Barton 2006).
The impact of fusions on the recombination landscape has received less attention – but evidence is emerging of substantial effects. Theory predicts that recombination suppression could also lock up potentially adaptive loci around the fusion site (Guerrero and Kirkpatrick 2014). Marin-Garcia et al. (2024) studied wild mice populations with multiple fusions combining cytological analyses of meiotic configurations with linkage disequilibrium (LD) analyses of SNPs. Recombination in metacentrics shifted towards telomeres relative to acrocentrics leading to reduced recombination rate genome-wide and increased genomic divergence. Major shifts in recombination rates between all acrocentric and multiple-fusion populations was concentrated in regions with immune system and olfaction genes. However, recombination rates shifted primarily in response to genotype at a key gene (Prdm9), with effects of fusions per se restricted to centromeric regions.
Adaptive advantage of chromosome rearrangements. White (1978) and other proponents of underdominance as a primary driver of chromosomal speciation suggested that newly arisen chromosome changes might have adaptive advantages that would mitigate reduced fertility in heterozygotes. This could happen via position effects, where gene expression is altered at or near the breakpoints, or positive epistasis between newly linked genes. One such example is the effect of a Robertsonian translocation on expression of genes associated with muscle development in the gayal (Bos frontalis) which lives in rugged mountains (Li et al. 2023). Otherwise, most evidence for positively selected rearrangements comes from analysis of large inversions that are segregating within species and are associated with ecological divergence (Berdan et al. 2023). High-resolution analyses of rearrangements in several species indicates that few breakpoints occur at boundaries of Topologically Associating Domains (TADs; see below), suggesting that most surviving changes have limited effect on gene expression. Across great apes and humans, deletions at TAD breakpoints are selected against, especially in association with strong transcription start sites (Fudenberg and Pollard 2019). Clearly, there is much more to be learnt here, especially as studies combining high resolution mapping of chromosome rearrangements and effects on gene expression are expanded to more systems.
Meiotic drive. As only one of four products of meiosis survive as a mature oocyte (the rest being relegated to polar bodies), variation in the strength of centromere interactions with the asymmetric spindle of the oocyte can cause specific forms of chromosomes to increase in frequency in a population. This is called meiotic drive and has long been recognised as a mechanism that could enable establishment of underdominant rearrangements (White 1978; Hedrick 1981; Walsh 1982). Across mammals, where Robertsonian fusions or centric shifts are common, karyotypes of most species are mostly acrocentric or metacentric, with a marked deficit of species with intermediate states (Pardo-Manuel de Villena and Sapienza 2001). This pattern is attributed to meiotic drive in which either metacentric or acrocentric morphologies are favoured, combined with frequent shifts in the direction of drive. When the direction of drive does change, it is expected that the rate of change in chromosome number will increase towards whatever state is now favoured. This prediction was supported using novel macroevolutionary models (Blackmon et al. 2019). The meiotic drive hypothesis is supported by extensive experimental evidence in mice, humans and birds (Pardo-Manuel de Villena and Sapienza 2001) and is now well understood mechanistically (e. g. for mice, Akera et al. 2017).
Why do taxa differ in the rate and form of chromosome rearrangements? It has long been recognised that the form, as well as rate, of chromosome change differs at phylogenetic scale. This is evident in the data from Bush et al. (1977) and was termed karyotypic orthoselection by White (1973; see also King 1993). The hope is that our rapidly increasing knowledge of genomic characteristics of evolutionary breakpoint regions (EBRs) and of chromatin architecture during interphase can shed some light. EBRs are typically depleted in coding genes, occur between rather than within TADs, and in some, but not all (Arias-Sardia et al. 2023) cases, are associated with expansions of repeated sequences including Transposable Elements (TEs; Yin et al. 2021; reviewed in Brannan et al. 2024). That mobilisation of TEs can lead to bursts of chromosome rearrangement is a long-standing hypothesis (Lawson et al. 2023) and there are now some clear examples relating expansions of particular TEs to elevated rates of chromosome reorganisation in mammals (O’Neill et al. 1998 in Macropods; Carbonne et al. 2014 for Gibbons; Sotero-Caio et al. 2015 in Phyllostomatid bats), presumably via non-homologous recombination. In the case of the Gibbons, insertions of the novel retrovirus were also shown to modify expression of genes controlling chromosome segregation.
These above examples aside, consistent differences in characteristics of EBRs between taxa with high rates of interchromosomal vs intrachromosomal rearrangements have proved elusive (Brannan et al. 2024). An alternative hypothesis – the Integrative Breakage Model (Farre et al. 2015) posits that propensity towards different forms of rearrangement relates to how chromatin is organised during interphase of germ-line cells. Álvarez-Gonzalez et al. (2022) explored this hypothesis using new evidence on 3D genome organisation (e. g. via HiC) across therian mammals. Eutherians have chromosomes organised in spatially distinct territories regardless of chromosome number whereas marsupials have clustered (“Rabl” form) centromeres. The resulting increased opportunity for inter-chromosomal interactions of chromatin territories in eutherians could explain higher rate of interchromosomal rearrangements. Marsupials have fewer, larger chromosomes with more intra-chrom rearrangements. No doubt, there is much more to learn here via integrated analyses of genome structure, chromatin organisation and gene expression across related species (Figure 3; Mohan et al. 2024).
Moving forward. Advances in genomic sequencing technologies now enable us to identify finer-scale structural variation (e. g., Figure 2) and we can now see that structural variation can occur at different scales, with different consequences of pre- or post-zygotic incompatibilities resulting in speciation (Berdan et al. 2023). But it is only with a multi-omics approach (e. g., chromatin structure, transcriptomics and epigenomics) that we will be able to piece together the consequences and mechanisms of structural variation evolution and outcomes on fitness/divergence (Figure 3). As well as taking this broad omics approach, we need to be mindful about the proposed connections to population-level processes of recombination, selection and drift. As recent works on model systems demonstrate, there is also exciting potential to scale from experimental manipulations at cellular scale to organismal and population processes (Ansai and Kitano 2022). As Bush et al. (1977) recognised, the greater the comparative breadth of insight we have across mammals, the more we can learn about what drives chromosome change and how this promotes organismal diversity.
Fortunately, with new theory and tools, the questions about why vertebrate taxa differ in rates and forms of chromosome change, and how all this relates to speciation remain hot topics. Classical cytogenetics left us with a rich diversity of systems to explore and it is now possible to compare genomes, chromatin organisation, gene expression and historical demography across such taxa. Bush et al. (1977) was a great start and there is much left for Jim Patton to ponder!
Literature cited
Akera, T., et al. 2017. Spindle asymmetry drives non-Mendelian chromosome segregation. Science 358:668-672.
Álvarez-González, L., et al. 2022. Principles of 3D chromosome folding and evolutionary genome reshuffling in mammals. Cell Reports, 41L 111839
Ansai, S. and Kitano, J. 2022. Speciation and adaptation research meets genome editing. Philosophical Transactions of the Royal Society of London B 377:20200516
Aria-Sardá, C., S. Quigley, and M. Farré. 2023. Patterns of chromosome evolution in ruminants. Molecular Ecology DOI: 10.1111/mec.17197.
Ashley, T. 2002. X-autosome translocations, meiotic synapsis, chromosome evolution and speciation. Cytogenetics and Cell Genetics 96:33-39
Baker, R. J., and J. W. Bickham. 1986. Speciation by monobrachial centric fusions. Proceedings of the National Academy of Sciences 83:8245-8248.
Berdan, E. L., et al. 2023. How chromosomal inversions reorient the evolutionary process. Journal of Evolutionary Biology 36:1761-1782.
Blackmon, H., et al. 2019. Meiotic drive shapes rates of karyotype evolution in mammals. Evolution 73:511-523.
Brannan, E. O., G. A. Hartley, and R. J. O’Neill. 2024. Mechanisms of rapid karyotype evolution in mammals. Genes 15:62-85.
Bredeson, J. V., et al. 2024. Conserved chromatin and repetitive regions reveal slow genome evolution in frogs. Nature Communications 15:579.
Bush, G. L., et al. 1977. Rapid speciation and chromosomal evolution in mammals. Proceedings of the National Academy of Sciences 74:3942-3946.
Carbone, L., et al. 2014. Gibbon genome and the fast karyotype evolution of small apes. Nature 513:195-201.
Close, R. L., et al. 1996. Spermatogenesis and synaptonemal complexes of hybrid Petrogale (Marsupialia). Journal of Heredity 87:96–107.
Coyne, J. A., and H. A. Orr. 2004. Speciation. Oxford University Press Inc.
Damas, J., M. Corbo, and H. A. Lewin. 2021. Vertebrate chromosome evolution. Annual Review of Animal Biosciences 9:1-27.
Eldridge, M. D. B., and R. L. Close. 1992. Taxonomy of rock wallabies, Petrogale (Marsupialia, Macropodidae). 1. A revision of the Eastern Petrogale with the description of 3 new species. Australian Journal of Zoology, 40:605-625.
Faria, R., et al. 2018. Evolving inversions. Trends in Ecology and Evolution 34:239-248.
Farre, M. et al. 2012. Recombination rates and genomic shuffling in human and chimpanzee - a new twist on the chromosomal speciation theory. Molecular Biology and Evolution 30:853-864
Farre M. et al. 2015. An integrative breakage model of genome architecture, reshuffling and evolution. Bioessays 37: 479-488.
Fudenberg, G., and K. S., Pollard. 2019. Chromatin features constrain structural variation across evolutionary timescales. Proceedings of the National Academy of Sciences 116:2175-2180.
Guerrero, R. F., and M. Kirkpatrick. 2014. Local adaptation and the evolution of chromosome fusions. Evolution 68:2747-2756.
Hager, E. R., et al. 2022. A chromosomal inversion contributes to divergence in multiple traits between deer mouse ecotypes. Science 377:399-405.
Haldane, J. B. S. 1922. Sex ratio and unisexual sterility in hybrid animals. Journal of Genetics 12:101–109.
Hedrick, P. W. 1981. The establishment of chromosomal variants. Evolution 35:322–332.
Herrick, J., and B. Sclavi. 2019. Genome diversity and species richness in mammals. bioRxiv 709311.
Jonicka, M. M. et al. 2024. Drift drives the evolution of chromosome number II: the impact of range size on genome evolution in Carnivora. Journal of Heredity esae025.
King, M. 1993. Species Evolution: The Role of Chromosome Change. Cambridge: Cambridge University Press.
Kirkpatrick, M., and N. Barton. 2006. Chromosome inversions, local adaptation and speciation. Genetics 173:419-434.
Kopania, E. E., et. al. 2022. The contribution of sex chromosome conflict to disrupted spermatogenesis in hybrid house mice. Genetics 222: iyac151.
Kronenberg, Z. N., et. al. 2018. High-resolution comparative analysis of great ape genomes. Science 360: eaar6343.
Levin, D. A., and A. C. Wilson. 1976. Rates of evolution in seed plants: net increase in diversity of chromosome numbers and species numbers through time. Proceedings of the National Academy of Sciences 73:2086-2090.
Lawson, H. A. et al. 2023. Transposable elements in mammalian chromatin organisation. Nature Reviews Genetics 24: 712-723
Li, Y., et al. 2023. Large-scale chromosomal changes lead to genome-level expression alterations, environmental adaptation, and speciation in the Gayal (Bos frontalis). Molecular Biology and Evolution 40:msad006.
Lucek, K., et al. 2023. The impact of chromosomal rearrangements in speciation: from micro- to macroevolution. Cold Spring Harbor Perspectives in Biology 15:a041447.
Marín-García, C., et al. 2024. Multiple genomic landscapes of recombination and genomic divergence in wild populations of house mouse – the role of chromosomal fusions and Prdm9. Molecular Biology and Evolution 41:msae063.
Marta Farré, M., T. J. Robinson, and A. Ruiz-Herrera. 2015. An integrative breakage model of genome architecture, reshuffling and evolution. Bioessays 37:479-488.
Martinez, P. A., et. al. 2017. A comparative study on karyotypic diversification rate in mammals. Heredity 118:366-373.
Mohan, A. V., et al. 2024. A three-dimensional genomics view for speciation research. Trends in Genetics 40:638–641.
Muffato, M. et al. 2023. Reconstruction of hundreds of reference ancestral genomes across the eukaryotic kingdom. Nature Ecology and Evolution 7:355-366.
Navarro, A., and N. H. Barton. 2003. Chromosomal speciation and molecular divergence--accelerated evolution in rearranged chromosomes. Science 300:321-324.
O’Neill, R. J. W., M. J. O’Neill, and J. A. M. Marshall. 1998. Undermethylation associated with retroelement activation and chromosome remodelling in an interspecific mammalian hybrid. Nature 393:68-72.
Pardo-Manuel de Villena, F., and C. Sapienza. 2001. Female meiosis drives karyotypic evolution in mammals. Genetics 159:1179-1189.
Patton, J. L. 1967. Chromosome studies of certain pocket mice genus Perognathus (Rodentia: Heteromyidae). Journal of Mammalogy 48:27-37.
Patton, J. L., and S. W. Sherwood. 1983. Chromosome evolution and speciation in rodents. Annual Review of Ecology and Systematics 14:139-158.
Patton, J. L. 2004. Comparative genomics and the role of chromosomal rearrangements in species divergence: a paradigm revisited. Mastozoología Neotropical 11:147-150.
Potter, S., et al. 2022. Limited introgression between rock-wallabies with extensive chromosomal rearrangements. Molecular Biology and Evolution 39:msab333.
Sites Jr, J.W., and C. Moritz. 1987. Chromosomal evolution and speciation revisited. Systematic Zoology 36:153-174.
Sotero-Caio, C. G., et al. 2015. Integration of molecular cytogenetics, dated molecular phylogeny, and model-based predictions to understand the extreme chromosome reorganization in the Neotropical genus Tonatia (Chiroptera: Phyllostomidae). BMC Evolutionary Biology 15:220.
Vara, C., et al. 2021. The impact of chromosomal fusions on 3D genome folding and recombination in the germ line. Nature Communications 12:2981.
Yin, Y. et al. 2021. Molecular mechanisms and topological consequences of drastic chromosomal rearrangements of muntjac deer. Nature Communications 12:6858.
Yu, H. et al. 2024. Pan-evolutionary and regulatory genome architecture delineated by an integrated macro- and microsynteny approach. Nature Protocols 19:1623-1678.
Walsh, J. B. 1982. Rate of accumulation of reproductive isolation by chromosome rearrangements. The American Naturalist 120: 510-532.
Waters, P. D., et al. 2021. Microchromosomes are building blocks of bird, reptile, and mammal chromosomes. Proceedings of the National Academy of Sciences 118:e2112494118.
Wellenreuther, M., and L. Bernatchez. 2018. Eco-evolutionary genomics of chromosomal inversions. Trends in Ecology and Evolution 33:427-440.
Westerman, M., R. W. Meredith, and M. S. Springer. 2010. Cytogenetics meets phylogenetics: a review of karyotype evolution in diprotodontian marsupials. Journal of Heredity 101:690-702.
White, M. J. D. 1973. Animal cytogenetics and evolution. Cambridge University Press, Cambridge, UK.
White, M. J. D. 1978. Modes of speciation. W. H. Freeman. San Francisco, U.S.A.
Wright, S. 1931. Evolution in Mendelian populations. Genetics 16:97-159.
Wright, S. 1940. Breeding structure of populations in relation to speciation. American Naturalist 74:232-248.
Associated editors: Marjorie Matocq and Eileen Lacey
Submitted: September 19, 2024 Reviewed: December 9, 2024
Accepted: January 14, 2025; Published on line: January 31, 2025
_Speciation_vs_chromosome_arm_evolution_vertebrates.png)
Figure 1. Key results from Bush et al. (1977). i) Rates of change in chromosome number of extant mammalian and other vertebrate taxa against net speciation rate. ii) Rates of chromosome arm number evolution of extant mammalian and other vertebrate taxa against net speciation rate. The R2 statistics for these correlations are inset in each graph along with the trend lines for mammals (black dotted line) versus other vertebrates (grey dotted line).
i)
ii)
Figure 2. Expansion of types of genome rearrangements readily detected with direct assembly from long-read sequencing versus classical cytogenetics (excluding polytene chromosomes).
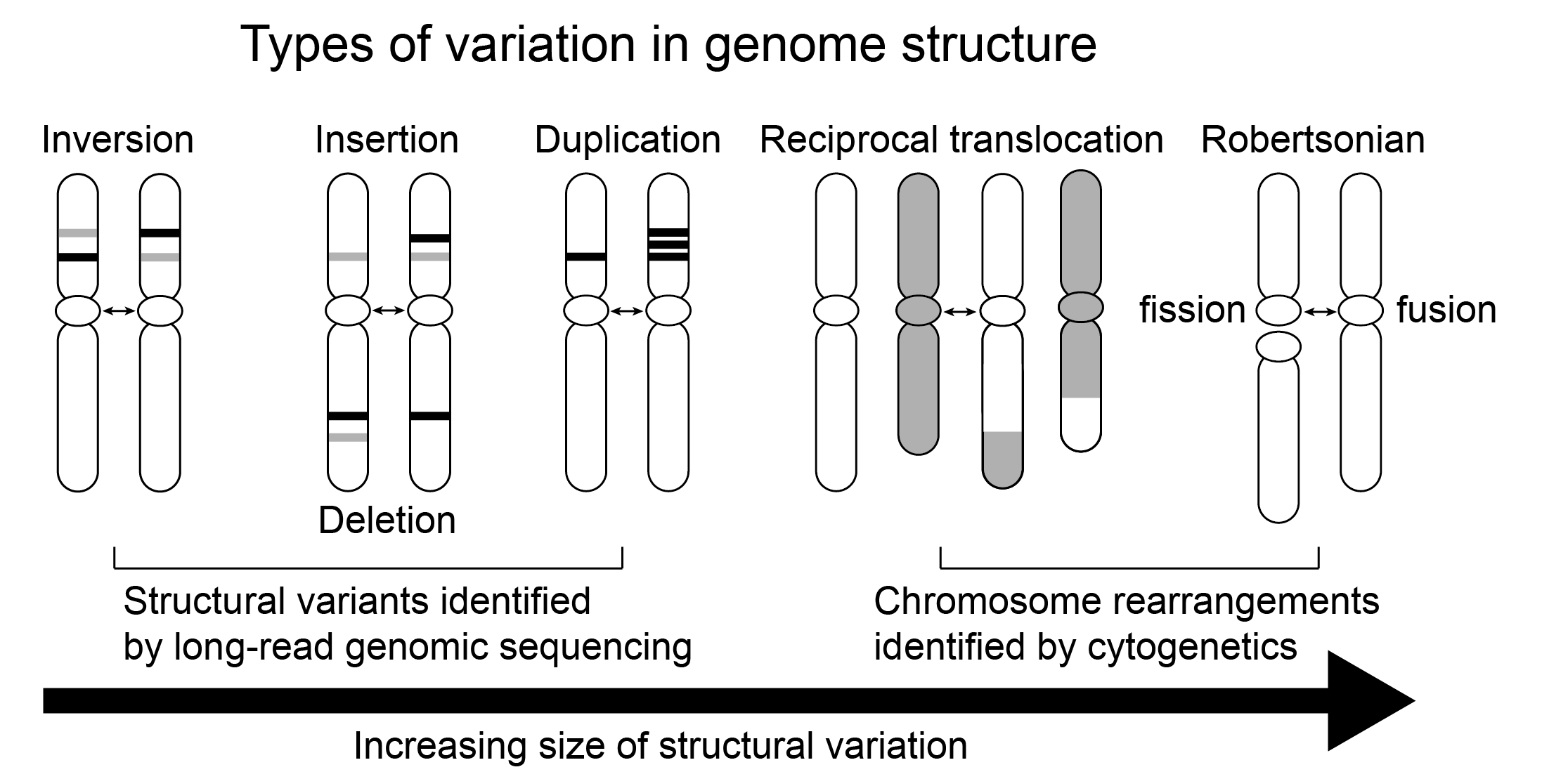
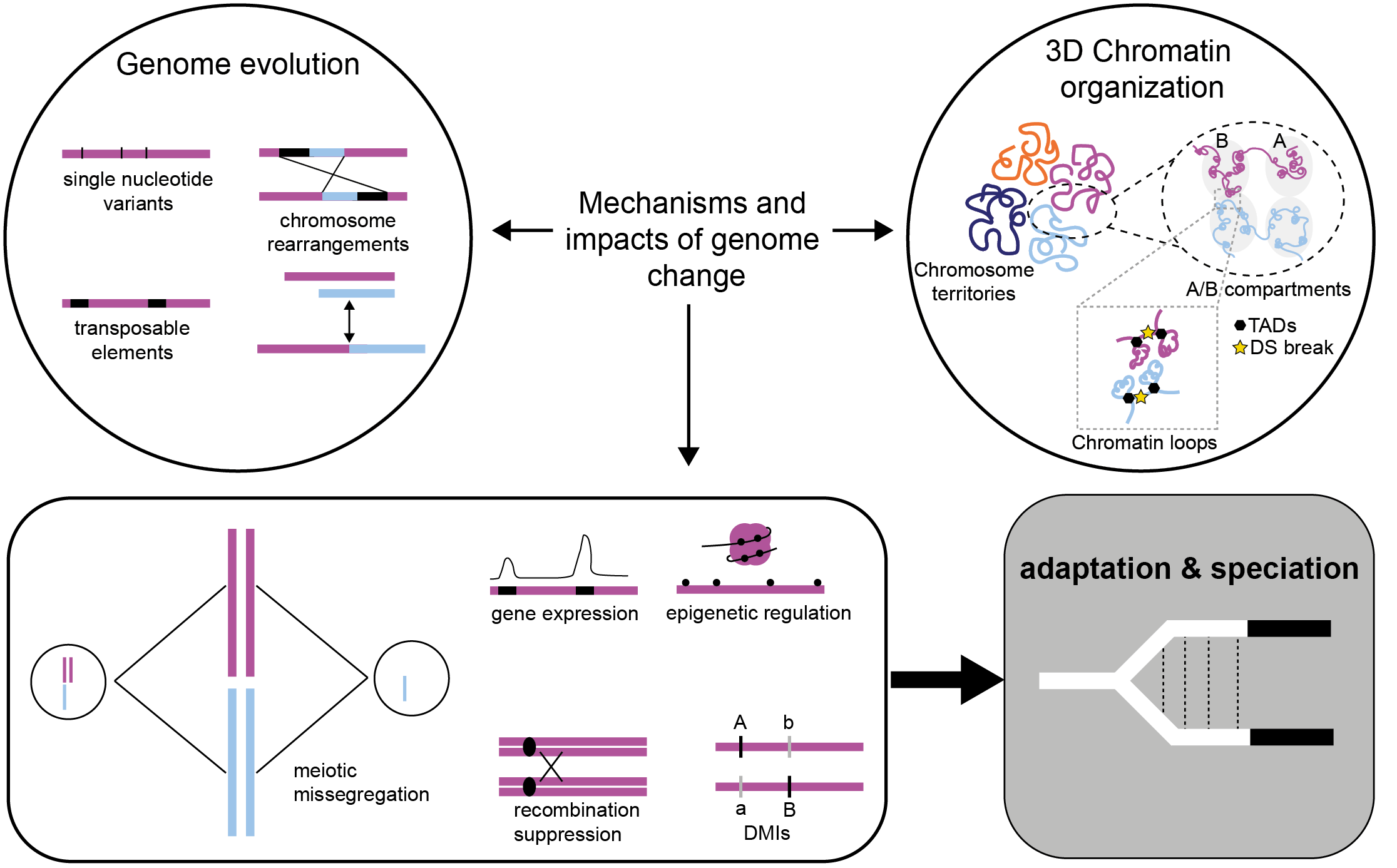
Figure 3. Integration of analyses of genome structure, chromatin organisation, gene function and recombination landscapes across closely related species is a promising new direction. Figure modified from Mohan et al. (2024)